Несовершенный остеогенез. Несовершенный остеогенез относится к редко встречающимся врожденным заболеваниям скелета. Различают 4 формы несовершенного остеогенеза: внутриутробную, детскую, юношескую и форму, поражающую людей в возрасте после периода полового созревания. Мы дадим описание лишь внутриутробной формы.
Этиология несовершенного остеогенеза не ясна. В литературе имеются многочисленные указания на семейный характер заболевания, встречающегося иногда в нескольких поколениях.
В основе несовершенного остеогенеза лежит врожденная недостаточность остеобластов, проявляющаяся в нарушении периостального остеогенеза. Эпифизарный хрящ длинных трубчатых костей не изменен, и поэтому рост кости в длину, исходящий из эпифизарного росткового хряща, протекает нормально. В то же время вследствие недостаточного периостального и энхондрального остеогенеза подавлен и изменен рост кости в ширину. Кость бедна костными элементами и имеет неправильное строение. Компактное вещество кости резко истончено и часто бывает не толще яичной скорлупы. В результате снижаются механические качества кости и создаются условия для патологической ее ломкости. Дети рождаются с чрезвычайно укороченными и деформированными конечностями. Имеется также недостаточное костеобразование и в черепе, вследствие чего он при ощупывании представляется мягким, имеющим множество брешей («каучуковая голова»).
Рентгенологическое исследование показывает, что укорочение длинных трубчатых костей и их деформация обусловлены множеством переломов, имевших место во внутриутробной жизни и часто неправильно сросшихся (см. рис. 75). В очень тяжелых случаях при ощупывании конечностей создается впечатление наполненного костными осколками мешка. В случаях средней тяжести число свежих переломов в конечностях меньше, но имеются множественные изгибы и искривления костей как результат имевших место внутриутробных переломов.
Характерными для несовершенного костеобразования являются нормальные размеры кистей и стоп; фаланги не ломаются никогда, пястные же и плюсневые кости лишь в исключительно редких случаях. Поэтому рядом с короткими неуклюжими толстыми проксимально расположенными костями пальцы производят впечатление несоразмерно длинных, как бы заостренных. Кожа, покрывающая укоротившиеся кости, ложится поперечными складками, хорошо определяемыми на рентгенограмме и нередко принимаемыми недостаточно опытными рентгенологами за линии переломов. В отличие от последних они наиболее отчетливо определяются вне кости на поверхности контуров мягких тканей. Позвоночник при внутриутробной форме несовершенного костеобразования, как правило, у новорожденного не изменен.
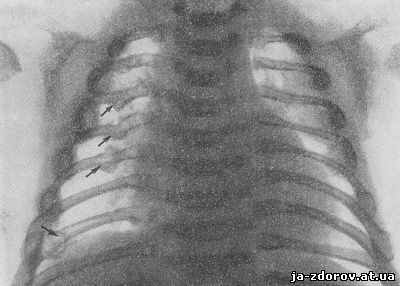
Рис. 74. Несовершенный остеогенез у новорожденного. Грудная клетка. Костные мозоли в области ребер.
В соответствии с уменьшенным содержанием в кости костных элементов и минеральных солей при рентгенологическом исследовании определяется также повышенная прозрачность костей; компактный слой резко истончен, а костномозговой канал эксцентрически увеличен в диаметре и местами неровен. Губчатая структура часто разрежена, нередко она вовсе не определяется. На первый взгляд рентгенограммы скелета производят впечатление технически неудачных. Иногда на рентгенограмме почти невозможно отличить тень кости от тени мягких тканей. Помимо множественных переломов длинных трубчатых костей с изгибами, определяются также переломы ребер без больших смещений с костными узловатыми мозолями; иногда на одном ребре определяется несколько костных мозолей (рис. 74, 75).
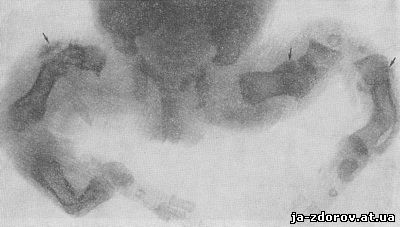
Рис. 75. Несовершенный остеогенез у новорожденного. Нижние конечности.
На рентгенограмме черепа отмечается резкое истончение костей свода. Лучше обызвествлена затылочная кость (рис. 76). На фоне тени свода черепа иногда видны отдельные вкрапления или различной формы островки окостенения, придающие черепу мозаичный рисунок. Швы и роднички расширены.
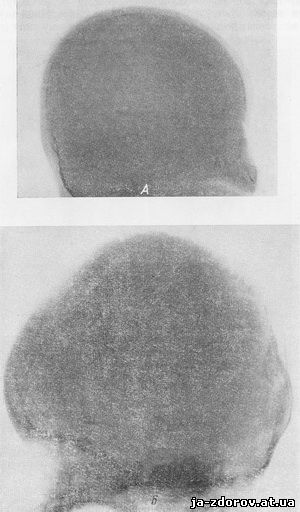
Рис. 76. Череп новорожденного.
А - каучуковый мешок; Б - мозаичный рисунок костей свода.
Распознавание этого заболевания обычно не представляет трудностей.
Прогноз при внутриутробной форме несовершенного остеогенеза плохой. Дети редко рождаются живыми или же они умирают в первые дни жизни. Однако у выживших детей в дальнейшем может наступить улучшение костеобразования; переломы в этих случаях происходят реже, но вследствие наличия плохо излеченных переломов дети, как правило, остаются калеками.
При внешнем осмотре детей с врожденным несовершенным остеогенезом бросается в глаза чрезвычайная голубизна склер.
Легкая форма врожденного несовершенного остеогенеза получила название остеопсатироза. Первые переломы в этих случаях происходят не внутриутробно, а во время родов или непосредственно после родов (рис. 77), часто же спустя несколько недель и даже месяцев после рождения. С. А. Рейнберг относит остеопсатироз к детской форме несовершенного остеогенеза.
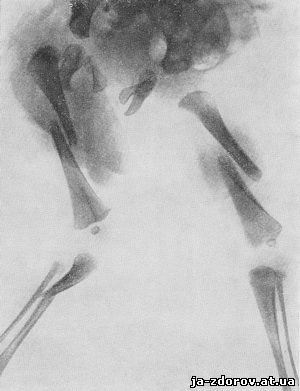
Рис. 77. Остеопсатироз у ребенка 1 суток. Переломы бедренных костей и костей голени. Вскрытие. Диагноз подтвержден гистологически.
Фетальная хондродистрофия. Фетальная хондродистрофия была описана в 1876 г. Parrot, в 1892 г. Kaufmann и в 1900 г. Pierre-Marie. Ее называют также ахондроплазией, хондромаляиией, микромелией. Это заболевание характеризуется нарушением процессов энхондрального окостенения преимущественно в эпифизарных концах длинных трубчатых костей, в результате чего получается укорочение конечностей. Периостальный же и эндостальный рост кости в толщину протекает нормально. Таким образом, фетальная хондродистрофия ведет к карликовому росту и представляет собой полную противоположность несовершенному остеогенезу. Поражаются только кости, развивающиеся по энхондральному типу; кости же, развивающиеся по соединительнотканному типу (плоские кости свода черепа, ключицы), поражаются в наиболее тяжелых случаях и лишь в незначительной степени.
Различают три формы фетальной хондродистрофии: гиперпластическую, гипопластическую и маляцическую, состоящую в размягчении хряща. Гистологически во всех случаях наблюдается беспорядочное расположение хрящевых клеток в ростковом хряще в отличие от нормальной картины расположения их ровными столбиками, а также неправильное обызвествление. Гиперпластическая форма характеризуется чрезмерным разрастанием и увеличением размеров эпифизарного хряща, гипопластическая - его уменьшением.
Изменения в костях имеют строго симметричный характер. В первую очередь поражаются кости конечностей; ребра же, кости таза и основание черепа вовлекаются лишь позднее. Меньше всего изменяются позвоночник, ключица и остальные кости туловища. В конечностях же в большей степени поражаются их проксимальные отделы (плечевая кость, бедренная); кости голени и предплечья укорочены в меньшей степени; еще меньше - плюсневые кости, пястные кости и фаланги.
В результате получается характерная картина непропорционального карликового роста, своеобразная микромелия: нормальные размеры туловища и позвоночника, короткие конечности и несколько увеличенная в размерах голова.
Различают два типа заболевания: «тип мопса» с изменениями, выраженными преимущественно в черепе, и «тип таксы» с изменениями, выраженными преимущественно в конечностях.
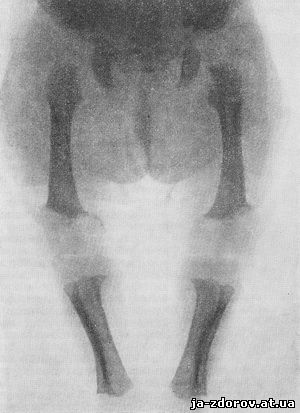
Рис. 78. Хондродистрофия нижних конечностей у ребенка 2 суток.
В рентгеновском изображении определяется резкое укорочение диафизов и, так как рост костей в толщину не нарушен, кость представляется широкой и как бы утолщенной (рис. 78). Компактный слой в средней части диафиза утолщен. При гиперпластической форме хондродистрофии зубчатый край метафизов часто грибовидно вздут и выступает за концы диафизов. На кисти все пальцы укорочены, имеют одинаковую длину (рис. 79), а второй, третий и четвертый пальцы, реже и пятый, расположены не параллельно друг другу, а лучеобразно расходятся, как бы образуя трезубец. Фаланги резко укорочены и имеют почти квадратную форму. В черепе определяется резкое выступание лобных бугров, основание его на боковом снимке представляется укороченным; скат клиновидной кости стоит круто; гипофизарная ямка уплощена и малых размеров. Плоские кости свода как бы нависают над лицевым черепом. Лопатки малых размеров, серповидной формы. Иногда наблюдаются уплощенные позвонки, межпозвонковые хрящи несколько большей высоты, чем в норме, клиновидная деформация одного или нескольких позвонков и в редких случаях генерализованная платиспондилия. В поясничном отделе определяется лордоз, а на границе грудного и поясничного отделов иногда угловой кифоз. Ребра по сравнению с другими костями укорочены меньше и расположены горизонтально; их грудинные концы, головки и шейки утолщены и деформированы. Часто наблюдается удлиненная малоберцовая кость.
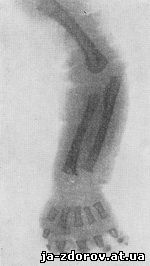
Рис. 79. Хондродистрофия у новорожденного (рентгенограмма ручки).
Остеохондродистрофия. Это системное не столь редкое заболевание имеет множество других названий: генерализованная остеохондропатия с симметричной гипоплазией суставов, системная остеохондропатия, множественная эпифизарная дисплазия, множественная местная маляция, платибрахиспондилия, атипическая хондродистрофия и др.
Остеохондродистрофия иногда имеет семейный характер, когда в одной семье поражаются несколько членов в одном, двух или больше поколениях. При внешнем осмотре бросается в глаза малый рост, обусловленный укорочением туловища, причем позвоночный столб, а вместе с ним все туловище укорачивается на одну треть и даже вдвое против нормы. Конечности не укорачиваются и по сравнению с коротким туловищем производят впечатление непропорционально длинных. Нормальная по размерам голова как бы «сидит» на плечах вследствие укорочения шейного отдела позвоночника.
Этиология и патогенез остеохондродистрофии не выяснены. Сущность же ее состоит в глубоком врожденном нарушении процессов превращения хряща в костную ткань, т. е. в своеобразной неполноценности энхондрального, но не периостального окостенения. В то время как у новорожденного с хондродистрофией вся основная патология уже имеется налицо, при остеохондродистрофии патология развивается, как правило, лишь постепенно и начинает выявляться лишь к 5 - 6 годам.
Черепно-ключичный дизостоз. Эта аномалия, имеющая выраженный семейный характер, была впервые описана в 1883 г. и подробнее исследована в 1897 г. Marie и Sainton вскоре после открытия рентгеновых лучей. Сущность ее заключается в расстройстве процессов окостенения костей черепа и грудной клетки соединительнотканного происхождения. Она характеризуется прежде всего полным отсутствием или недоразвитием ключиц и грудины.
Чаще отсутствует только наружный конец одной или обеих ключиц, реже грудинный. Иногда ключица состоит из двух половин, соединенных фиброзным тяжом. Различные варианты частичного отсутствия или недоразвития ключиц могут быть односторонними или двусторонними. Иногда отсутствует грудина. Другой особенностью врожденного черепно-ключичного дизостоза являются изменения черепа: широко зияют швы; крыша черепа в значительной своей части остается в соединительнотканной фазе развития, не содержит известковых солей и не окостеневает. В то же время из центров плоских костей исходят островки костной ткани, имеющие вид раковин. Мозговой череп увеличен в поперечнике и уменьшен в передне-заднем направлении; лицевой череп уменьшен за счет гипоплазии верхних челюстей. Почти всегда уменьшены размеры туловища.
При черепно-ключичном дизостозе в других отделах скелета также часто определяются аномалии развития: уменьшение размеров лопатки, соха vara, соха valga, вывих бедра, задержка в развитии лобковых костей с наличием широкого симфиза, задержка в появлении ядер окостенения в конечностях, укорочение конечных фаланг пальцев кистей и стоп и др.
Мы наблюдали одного ребенка с черепно-ключичным дизостозом. Ребенок одних суток был направлен к нам для рентгенологического исследования по поводу предполагавшегося родового повреждения плечевого пояса. На снимке грудной клетки и плечевого пояса были обнаружены рудименты левой ключицы и полное отсутствие правой. На дополнительно произведенной боковой рентгенограмме грудной клетки мы установили отсутствие ядер окостенения грудины (рис. 80). На рентгенограммах черепа определялись задержка окостенения костей свода, широкие швы и роднички (рис. 81).
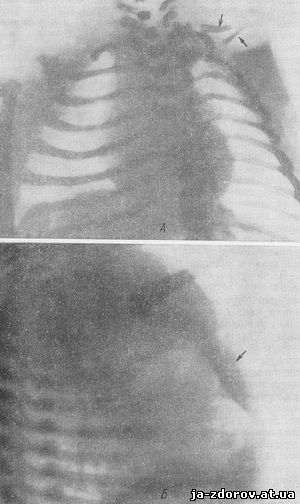
Рис. 80. Черенно-ключичный дизостоз у ребенка 1 суток (рентгенограммы грудной клетки).
А - в задней проекции; рудименты левой ключицы указаны стрелками; Б - в боковой проекции; в грудине отсутствуют ядра окостенения (указано стрелками).
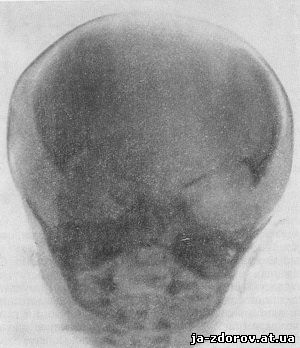
Рис. 81. Рентгенограмма головы того же ребенка, что и на рис. 80.
Недоразвитие костей черепа. Широкие швы и большие роднички.
Нижнечелюстно-лицевой дизостоз Бери - представляет собой аномалию черепа, сочетающуюся с синдактилией и с аномалиями позвоночника и других отделов скелета. Характеризуется гипоплазией костей лицевого черепа: скуловых костей, нижней челюсти, височной и других костей. Наблюдается также гипоплазия уха (ушной раковины, среднего и внутреннего уха), асимметричная конфигурация черепа, круто спускающееся спереди назад основание черепа, расположение глазных щелей с направлением их осей книзу и кнаружи, отсутствие или резкое уплощение носолобного угла.
Черепно-лицевой дизостоз. Эта врожденная аномалия, передающаяся по наследству, характеризуется следующей триадой: башенный череп в результате множественных синостозов черепных швов, преимущественно коронарного и ламбдовидного; внутренняя гидроцефалия и гипоплазия нижней челюсти (рис. 82). Наблюдается уплощенное дно глазниц, предрасполагающее к люксации глазных яблок книзу, экзофтальм и гипертелоризм (широко расставленные глаза).
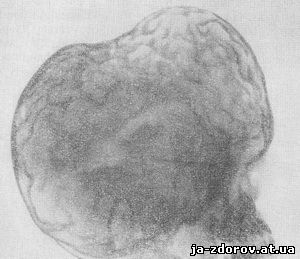
Рис. 82. Черепно-лицевой дизостоз у ребенка 1 суток (рентгенограмма по Catel).
Акроцефало-синдактилия Апера. Комбинированный порок развития черепа типа Апера характеризуется наличием краниостеноза с башенным черепом и сращением пальцев кистей и стоп, причем в одних случаях имеется сращение только в мягких тканях, в других - костный синостоз. Характерна форма лица с широким корнем носа. При рентгеновском исследовании помимо краниостеноза и синостоза пальцев на боковой рентгенограмме определяется круто ниспадающее спереди назад основание черепа.
Мелореостоз. Это врожденное заболевание скелета выражается в остеосклерозе одной только конечности, развивающемся вдоль одной ее стороны, в виде слегка волнистой полосы, переходящей через линию суставов с одной кости на другие кости той же конечности. В результате может оказаться пораженной целая верхняя или нижняя конечность. Эти полосы склероза сравнивают со стекающим и застывающим вдоль свечи стеарином или воском, откуда и возникло название «мелорестоз», что означает «стекающая вдоль конечности кость».
Поражения ребер не описаны. Корковый слой и эпифизы трубчатых костей, а также мелкие губчатые кости приобретают плотность слоновой кости; при этом корковый слой трубчатых костей утолщается в сторону костномозгового канала, суживая его, а также кнаружи, увеличивая наружный диаметр кости. Костная ткань, граничащая со склеротическими полосами, сохраняет нормальный рисунок. Этиология заболевания неизвестна.
Врожденная мраморная болезнь. Врожденная мраморная болезнь впервые описана в 1904 г. Albers-Schonberg. Она описана также под названием остеопетроза (окаменелости) костей. Этиология мраморной болезни не выяснена. В основе заболевания лежит нарушение процесса окостенения, выражающееся в том, что вырабатывается увеличенное количество компактного костного вещества на месте эндостального и энхондрального роста кости. В кости теряется нормальное разграничение между компактными губчатым веществом. Корковое вещество в два-три раза толще, чем в норме, причем утолщение его происходит почти целиком в сторону костномозгового канала, вследствие чего наступает сужение и даже запустевание костномозгового канала. В зоне предварительного обызвествления происходит чрезмерное отложение извести, сама зона расширяется. Точное распознавание врожденной мраморной болезни требует применения рентгеновского исследования, так как клинические ее проявления не всегда указывают на поражение скелета.
При рентгенологическом исследовании обнаруживается, что форма и размеры костей не изменены, но вся кость представляется однородной и компактной; костномозговой канал отсутствует или едва намечается. В диафизе трубчатых костей определяются гладкие периостальные наложения костной ткани, которые, в отличие от наложений при периоститах, не отделяются от компактного слоя кости костномозговым пространством. В то время как у взрослых поражаются равномерно почти все кости и спонгиозная масса в них замещается компактным веществом, у новорожденных и у маленьких детей вначале определяются поперечные полоски уплотнения кости шириной в несколько миллиметров в дистальном конце диафиза бедра и в проксимальном конце большеберцовой кости, а также в телах позвонков, реже в проксимальном конце диафиза плеча. Патогномоничным является развитие костного склероза от эпифизарного хряща в направлении к диафизу, что в рентгеновском изображении получает свое отражение в виде ряда чередующихся участков уплотнения и участков нормальной кости, расположенных параллельно эпифизарной линии. В редких случаях определяется продольная осевая слоистость, с полосами различной ширины.
В области дистальных концов бедер и большеберцовых костей и проксимальных концов плечевых костей наблюдаются булавовидные и бутылочновидные вздутия костей. Также поражаются череп и таз, в которых рентгенологически определяются очаги остеосклероза. Частым осложнением при мраморной болезни являются патологические переломы костей.
Остеопойкилия. Остеопойкилия впервые была описана в 1905 г. Stieda. Это врожденное системное заболевание скелета называют также врожденной рассеянной склерозирующей остеопатией или врожденной пятнистой множественной остеопатией. Заболевание выражается в том, что во всех костях, за исключением ключицы, в губчатом веществе оказываются включенными круглые и овальные островки плотной склерозированной ткани величиной от 2 до 10 мм. Поражаются больше всего кости запястья и предплюсны, а также прилегающие к ним эпифизарные концы длинных трубчатых костей, в меньшей степени метафизы; диафизы остаются непораженными, но отдельные островки склероза определяются и в диафизах длинных трубчатых костей. Островки костного склероза обнаруживаются также в фалангах пальцев костей и стоп, в пястных и плюсневых костях, в головке и шейке бедра и в головке плечевой кости. Череп и ребра поражаются в очень редких случаях, также и позвонки, за исключением поясничных и крестцовых позвонков, которые могут поражаться вместе с костями таза.
По размерам очагов различают мелкоочаговую и крупноочаговую пойкилию. По количеству островков уплотнения она может быть редкой и густой.
Описанные выше изменения в костях не сопровождаются какими-либо клиническими проявлениями, поэтому заболевание определяется случайно при рентгенологическом исследовании, произведенном по другому поводу. Гистологические исследования Шморля показали, что островки склероза построены не по типу компактной ткани, а представляют собой уплотненное губчатое вещество.
Арахнодакталия. Арахнодактилия была описана в 1896 г. Marfan, который назвал это системное заболевание скелета долихостеномелией. Aschard же в 1902 г. дал этому заболеванию название арахнодактилии, которое и стало в литературе общепризнанным.
Это редкое системное заболевание возникает во внутриутробной жизни, а при появлении ребенка на свет оно уже отчетливо выражено. Характерным для него является ненормальный рост в длину всех трубчатых костей, в особенности же фаланг кистей и стоп, а также пястных и плюсневых костей. Изменения обнаруживаются также в черепе и скелете туловища.
Наиболее важными клиническими признаками этого заболевания являются общая мышечная атрофия с атонией всей скелетной мускулатуры, недоразвитие подкожного жира вплоть до его отсутствия и необычайная дряблость связочного аппарата, что обусловливает резкое ограничение самостоятельных движений. Прогноз неблагоприятный: большинство детей умирает в первые годы жизни от любого привходящего заболевания. Этиология арахнодактилии неизвестна. При рентгенологическом исследовании обращает на себя внимание диспропорция в форме коротких и длинных трубчатых костей - увеличение размеров в длину и значительное уменьшение их в толщину. Корковый слой тонкий, в губчатом веществе количество трабекул уменьшено, сами трабекулы истончены. Получается картина атрофии костей с выраженным остеопорозом. Кости удлинены и истончены, особенно фаланги, метакарпальные и метатарзальные кости.
Врожденная эпифизарная точечная дисплазия. Это сравнительно редкое заболевание. Наблюдается главным образом в раннем детском возрасте. Описаны также случаи внутриутробного поражения. Прогноз неблагоприятный.
На рентгенограммах скелета в эпифизах на месте нормальных одиночных ядер окостенения определяется множество мелких костных образований (рис. 83).
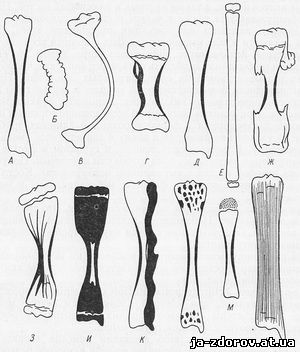
Рис. 83. Схематическое изображение рентгенологических проявлений деформации длинных трубчатых костей при различных врожденных системных заболеваниях скелета.
А - нормальная большеберцовая кость для сравнения; Б - несовершенное костеобразование; В - идиопатический остеопсатироз; Г - хондродистрофия; Д - остеохондродистрофия; Е - арахнодактилия; Ж - множественные хрящевые экзостозы; З - множественный хондроматоз костей; И - мраморная болезнь; К - мелореостоз; Л - остеопойкилия; М - врожденная эпифизарная точечная дисплазия; Н - врожденные системные диафизарные гиперостозы (по С. А. Рейнбергу).
Артрогрипоз. Врожденная аномалия развития, при которой наблюдаются резкие множественные контрактуры суставов верхних и нижних конечностей и дисплазия соответствующих групп мышц.
Клиническая картина этой аномалии описана впервые Отто в 1841 г. Главным образом обращает на себя внимание резкое ограничение движений в больших суставах. Суставы фиксированы в вытянутом или согнутом положении. Ранним симптомом является pes equino-varus. Кроме того, отмечается множество вывихов и подвывихов суставов.
Эта аномалия сочетается с кранио-фасциальными аномалиями, пороками сердца, аномалиями позвонков.
Рентгенологически обнаруживаются главным образом остеопороз костей, образующих пораженные суставы.
Врожденный энхондроматоз - болезнь Оллье. Врожденный энхондроматоз известен также под названиями «дисхондроплазия», «хондродисплазия», «множественный хондроматоз», «хондроматозная дисплазия», «болезнь Оллье» (описавшего эту болезнь в 1899 г.).
Заболевание считается врожденным. В метафизах длинных трубчатых костей и периферических участках плоских костей при этом заболевании костная ткань замещена гиалиновым хрящом. Чаще поражаются кости нижних конечностей. Череп, позвоночник и ключицы не поражаются.
На рентгенограмме определяются дугообразное искривление трубчатых костей и мелкие просветления различной величины и формы в области метафизов.
Врожденный диффузный генерализованный гиперостоз. Эта аномалия, описанная Koszewsky, проявляется у новорожденного тотчас после родов в виде мышечной гипертонии, повышения рефлексов и склонности к судорогам. При рентгеновском исследовании определяется диффузный гиперостоз костей всего скелета; структура костей сохранена; концы диафизов ровные с правильной границей. Анатомическая сущность процесса заключается в недостаточном рассасывании костной ткани (остеокластия, остеосклероз). Новорожденные умирают в первые же дни жизни.
Кортикальный гиперостоз «синдром Кэффи». В 1830 г. Roske сообщил о своем наблюдении над мальчиком грудного возраста, у которого он обнаружил субпериостальное новообразование костной ткани на обеих плечевых, лучевых и большеберцовых костях, а также на левой половине нижней челюсти. 4 аналогичных случая были опубликованы в 1945 г. Кэффи и Сильверманом, назвавшими это заболевание «детским кортикальным гиперостозом». В 1946 г. Смит, Поттер и Сильверман опубликовали еще 7 случаев. В литературе встречается и другое наименование: «синдром Кэффи» или «синдром Кэффи-Смита» (рис. 84).
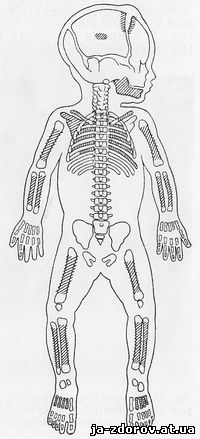
Рис. 84. Локализация гиперостозов при «синдроме Кэффи» (схема Catel).
Это своеобразное заболевание, этиология которого не выяснена, наблюдалось до сих пор только у грудных детей в возрасте менее шести месяцев. Свобода указывает, что болезнь может начаться еще во внутриутробной жизни.
Р. С. Левин (1957) наблюдал кортикальный гиперостоз у мальчика 3-недельного возраста, причем из анамнеза было выяснено, что припухлость на передних поверхностях голеней и на левом предплечье была обнаружена на 7-й день жизни. При пальпации отмечалась резкая болезненность согнутых в коленных суставах ножек. В клинику ребенок был направлен с диагнозом остеомиелита. При рентгенологическом исследовании был обнаружен кортикальный гиперостоз левой лучевой кости, бедренных костей и костей голени. При этом в области бедренных костей вновь образованная костная ткань была расположена не циркулярно, а лишь на передних и наружных поверхностях.
Болезнь протекает при повышенной температуре и ускоренной РОЭ. Поражаются все кости, кроме позвоночника, костей таза и мелких костей запястья и предплюсны; часто поражаются ключицы, наружные концы ребер, плечевая и локтевая кости; характерным считается поражение нижней челюсти. Поражения симметричные. В длинных трубчатых костях поражаются только диафизы. На рентгенограммах определяются резкий гиперостоз и остеосклероз, ведущие к значительной деформации костей и сужению костномозгового канала (рис. 85).
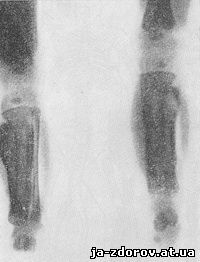
Рис. 85. Кортикальный гиперостоз у новорожденного. Рентгенограмма голеней. Резкое утолщение костей голени.
Наслоения новообразованной костной ткани располагаются кнаружи от кортикального слоя муфтообразно, иногда же только по одной или двум поверхностям пораженной кости. Наружные контуры вновь образованной кости четкие, слегка волнистые. Иногда во вновь образованной кости определяется продольная слоистость, которую рассматривают как выражение повторных обострений процесса.
Кортикальный гиперостоз наблюдается также в области лопаток и костях черепа. В мягких тканях соответственно изменениям в костях определяется припухлость и уплотнение. В течение года кость нормализуется, но возможны и остаточные явления; описаны случаи, когда все симптомы исчезли спустя 4 недели.
Р. С. Левин наблюдал кортикальный гиперостоз, длившийся 8 лет. Автор обращает внимание на то, что клинически гиперостоз проявляется не всегда и что клиническая картина по своей незначительной выраженности часто не соответствует распространенным и значительным костным изменениям, и заключает отсюда о важности рентгенологического исследования не только для распознавания заболевания, но и для уточнения распространения гиперостоза. Он указывает также, что клинические признаки заболевания исчезают значительно раньше, чем определяемые рентгенологически изменения.
При гистологическом исследовании было обнаружено утолщение надкостницы, от которой отходят плотные фиброзные тяжи по направлению к коже. Вновь образованная кость отслоена от нормального кортикального слоя кости; узкий костномозговой канал содержит костньш мозг. В мышечной ткани обнаруживаются дегенеративные изменения и фиброз; в артериях необычная пролиферация интимы. Воспаление и геморрагии не определялись.
При распознавании заболевания необходимо дифференцировать его от других гиперостозов и особенно от врожденного сифилиса.
Тератомы крестцово-копчиковой области относятся к дизонтогенетическим опухолям, которые развиваются на почве аномалий развития. Эти опухоли, содержащие эмбриональные ткани различной зрелости, иногда зачатки целых органов, различной величины и формы неоднородной консистенции (мягкой и твердой), внедряясь в полость малого таза, сдавливают смежные органы.
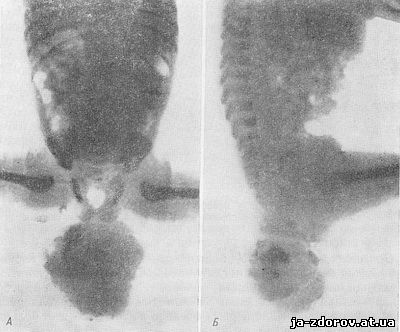
Рис. 86. Тератома крестцово-копчиковой области у ребенка 2 суток (рентгенограммы в двух проекциях - А и Б).
Рентгенограммы, произведенные в 2 проекциях, определяют характер опухоли, наличие известковых включений и элементов скелета (рис. 86). При дифференциальной диагностике со спинномозговой грыжей наличие или отсутствие изменений со стороны дужек соответствующего отдела позвоночника решает вопрос. При кистозной форме тератомы на рентгенограмме в пользу опухоли свидетельствует нормальная картина соответствующего отдела позвоночника.
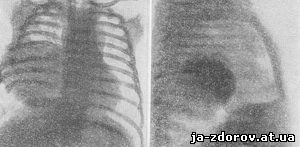
Рис. 87. Саркома ребра у новорожденного. Большая шаровидная опухоль между 5 ребром справа и диафрагмой (из книги Обернидермайра).
Остеогенная саркома. У новорожденных остеогенная саркома встречается очень редко. Иногда у новорожденного обнаруживается громадная опухоль конечности, области таза, позвоночника, ребра. Обнаруженная при рождении ребенка маленькая опухоль растет очень быстро и в течение первых месяцев жизни достигает больших размеров.
В случае, описанном Обернидермайром (1959), остеогенная саркома 5 ребра справа на рентгенограмме грудной клетки новорожденного имела вид огромной с резкими границами шаровидной опухоли, занимавшей значительную часть правой половины грудной клетки (рис. 87).
Типичные для остеогенной саркомы у взрослых и старших детей рентгенологические признаки у новорожденных не наблюдаются.















